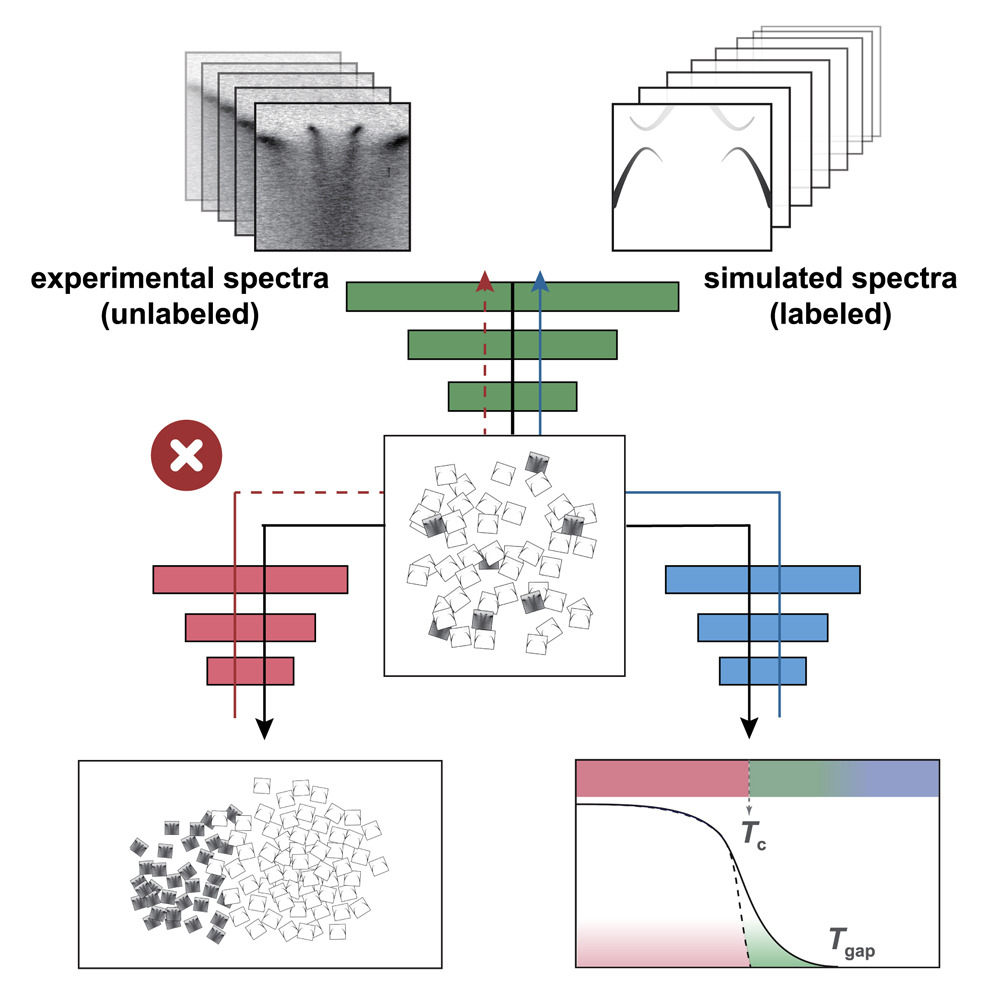
In correlated materials, strong quantum fluctuations hamper conventional spectral diagnosis of phase transitions, as the usual “energy gap” no longer serves as a reliable marker. Focusing on the superconductivity transition in such materials, this study employs machine-learning models to parse single-shot photoemission spectra and accurately pinpoint long-range superconducting order. The model recognizes subtle spectral cues invisible to gap-based analysis, unveiling the authentic transition even under heavy fluctuations. This approach enables rapid, non-contact detection of emergent phases in quantum materials, fueling fundamental exploration of strongly correlated systems and expediting the discovery of next-generation superconductors. Additionally, the authors introduce a domain-adversarial framework that merges large-scale simulation data with minimal experimental data, providing a robust solution to the persistent shortage of measurements in scientific machine learning. Together, these advances open new directions for understanding and harnessing complex quantum many-body states and phase transitions in quantum materials.
@article{chen2025detecting,title={Detecting thermodynamic phase transition via explainable machine learning of photoemission spectroscopy},author={Chen, Xu and Sun, Yuanjie and Hruska, Eugen and Dixit, Vivek and Yang, Jinming and He, Yu and Wang, Yao and Liu, Fang},journal={Newton},volume={1},number={3},year={2025},publisher={Elsevier},url={https://doi.org/10.1016/j.newton.2025.100066},preprint={https://doi.org/10.48550/arXiv.2406.04445},}
Automated Structural Refinement of Docked Complexes in Cytochrome P450 Using Molecular Dynamics
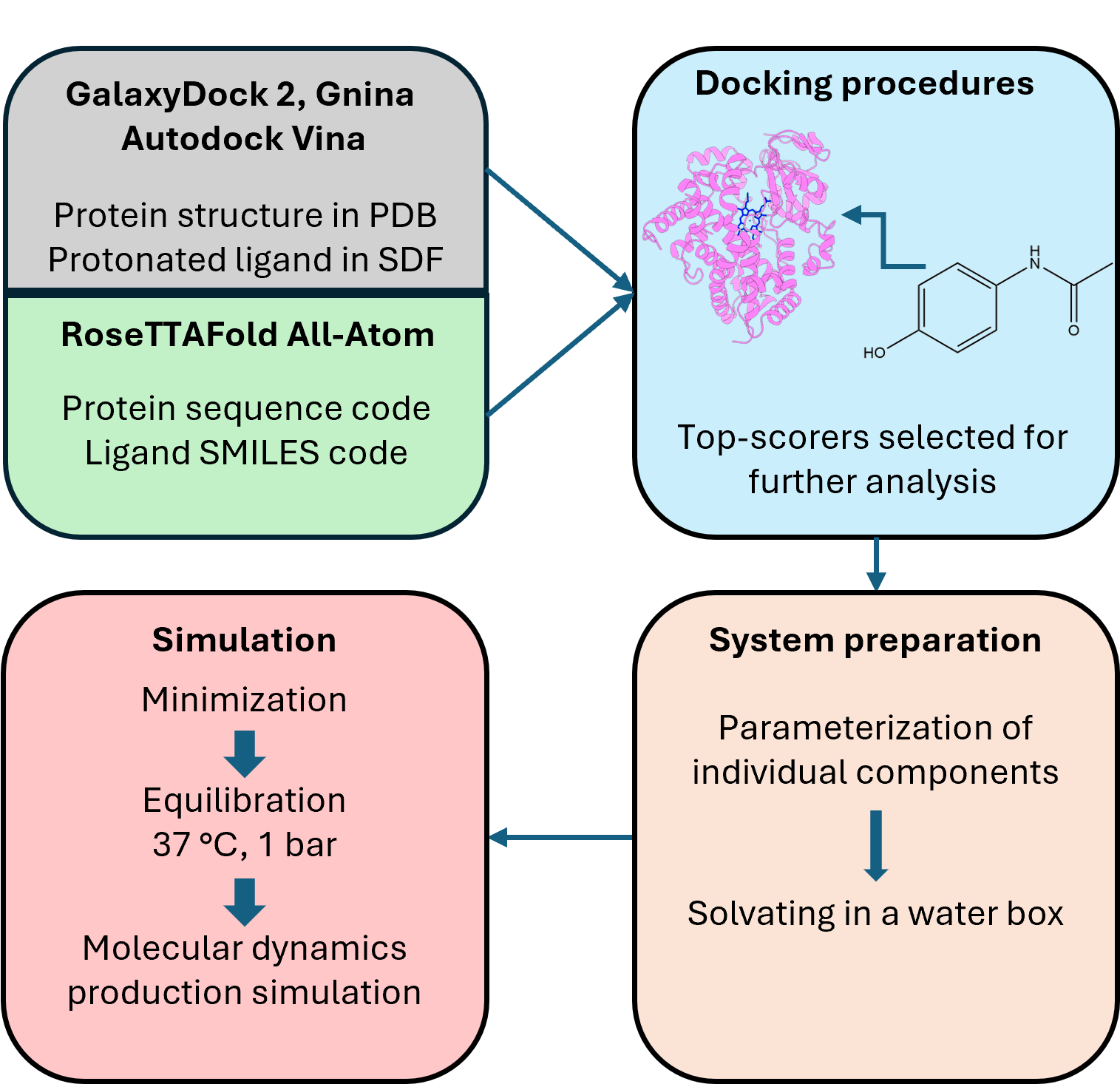
Docking and molecular dynamics are commonly used methods in drug development for the prediction of interactions between the drug in question and the target protein. When used together, docking studies can provide starting structures for molecular dynamics, which can improve the relatively low accuracy of docking but at greater computational cost. When the structure contains a cofactor with covalently bound metal atom, few automated methods are capable of setting up the system to perform molecular dynamics simulations. Another limitation that can be encountered is the parametrization of the ligand, which is not always easy to set up and automate using popular suites, such as AMBER or GROMACS. Here, we propose an automated workflow for setting up and running the simulation, providing a seamless integration for both flexible and rigid protein docking engines. The automatic setup works for around 90% of all ligands for rigid docking engines, with this number decreasing for RoseTTAFold All-Atom to around 30% due to its known problem with hallucination of the ligand conformation.
2024
The wins and failures of current docking methods tested on the flexible active site of cytochromes P450
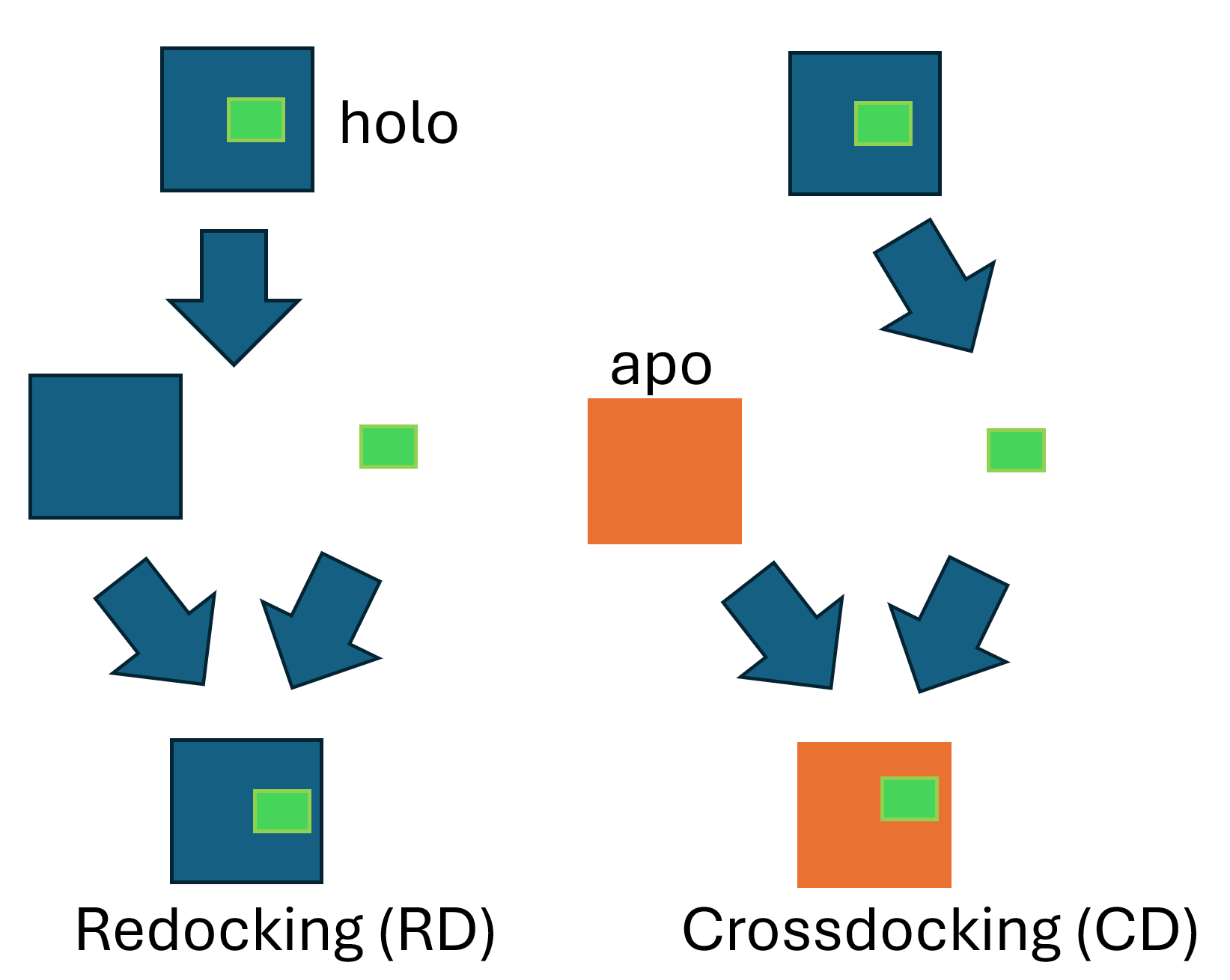
In this work, we benchmark 4 selected open source docking engines for use in the cytochrome P450 protein family. The key enzymes family of phase I metabolism is characterized by a wide variety of accepted substrates due to flexible active site. This work is a benchmark study which aims to evaluate the capabilities of current rigid and induced-fit docking methods for prediction of correct heme-ligand orientation. To asses it, we use two unique distances to heme iron and a SuCOS score to quantify reconstruction of orientation and chemical features. We selected three rigid protein docking engines: GNINA, AutoDock VINA, GalaxyDock2 HEME and a flexible docking model, RosettaFold-All-Atoms to test them on a dataset of 128 CYP-binding ligands. We report mean absolute error for RosetttaFold-All-Atom on key distance, to the atom closest to heme iron in experimental reference structure, 3 times lower than AutoDock VINA engine in the same simulation. Our results indicate that induced fit method is a significant improvement over rigid methods for flexible active site, but still offer limited predictivity. During crossdocking, RosettaFold-All-Atoms was able to recreate over a quarter of distances up to 20 percent difference from experiment. Further analysis indicates a low overlap in the distribution of ligand chemical features, based on a SuCOS score, which suggests a space for further improvement.
2022
Bridging the Experiment-Calculation Divide: Machine Learning Corrections to Redox Potential Calculations in Implicit and Explicit Solvent Models
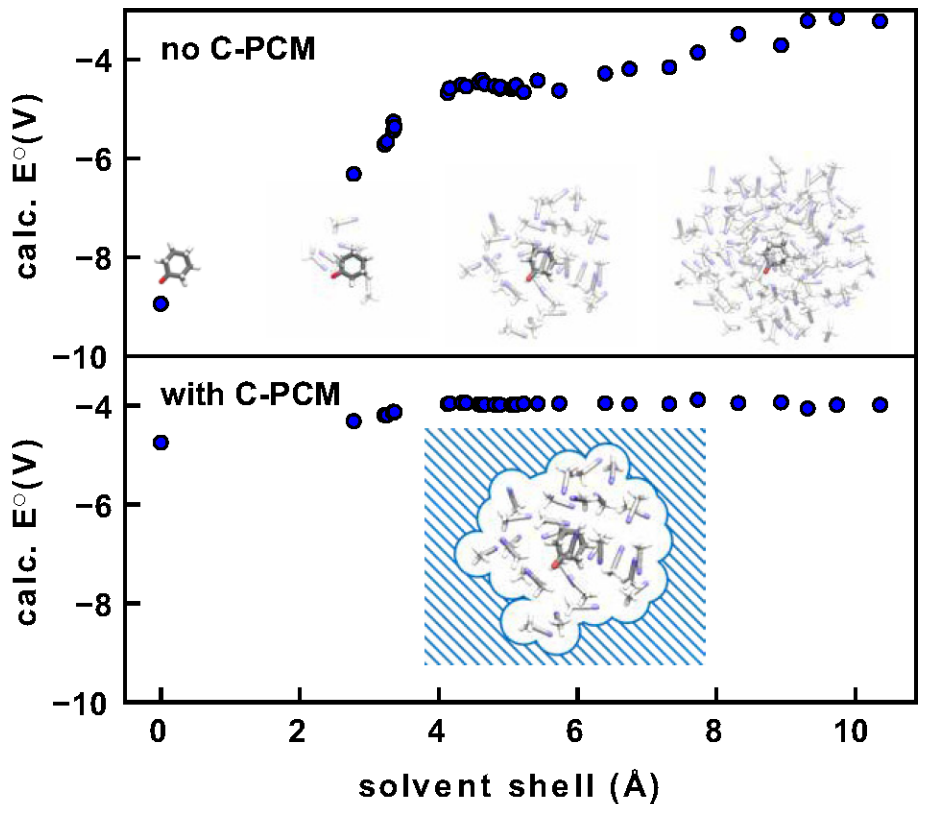
Prediction of redox potentials is essential for catalysis and energy storage. Although density functional theory (DFT) calculations have enabled rapid redox potential predictions for numerous compounds, prominent errors persist compared to experimental measurements. In this work, we develop machine learning (ML) models to reduce the errors of redox potential calculations in both implicit and explicit solvent models. Training and testing of the ML correction models are based on the diverse ROP313 data set with experimental redox potentials measured for organic and organometallic compounds in a variety of solvents. For the implicit solvent approach, our ML models can reduce both the systematic bias and the number of outliers. ML corrected redox potentials also demonstrate less sensitivity to DFT functional choice. For the explicit solvent approach, we significantly reduce the computational costs by embedding the microsolvated cluster in implicit bulk solvent, obtaining converged redox potential results with a smaller solvation shell. This combined implicit–explicit solvent model, together with GPU-accelerated quantum chemistry methods, enabled rapid generation of a large data set of explicit-solvent-calculated redox potentials for 165 organic compounds, allowing detailed investigation of the error sources in explicit solvent redox potential calculations.
@article{redox1,title={{Bridging the Experiment-Calculation Divide: Machine Learning Corrections to Redox Potential Calculations in Implicit and Explicit Solvent Models}},url={https://doi.org/10.1021/acs.jctc.1c01040},doi={10.1021/acs.jctc.1c01040},journal={J. Chem. Theory Comput.},author={Hruska, Eugen and Gale, Ariel and Liu, Fang},year={2022},preprint={https://chemrxiv.org/engage/chemrxiv/article-details/615f41128b620d134647d8c2},}
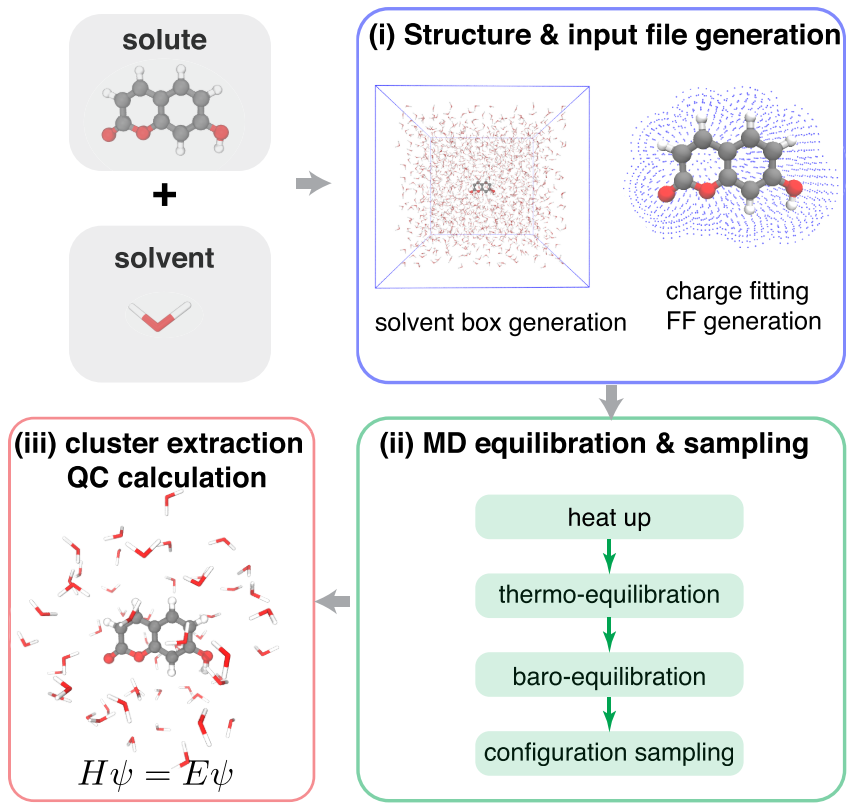
AutoSolvate : A Toolkit for Automating Quantum Chemistry Design and Discovery of Solvated Molecules
Eugen Hruska, Ariel Gale, Xiao Huang, and
1 more author
The availability of large, high-quality data sets is crucial for artificial intelligence design and discovery in chemistry. Despite the essential roles of solvents in chemistry, the rapid computational data set generation of solution-phase molecular properties at the quantum mechanical level of theory was previously hampered by the complicated simulation procedure. Software toolkits that can automate the procedure to set up high-throughput explicit-solvent quantum chemistry (QC) calculations for arbitrary solutes and solvents in an open-source framework are still lacking. We developed AutoSolvate, an open-source toolkit to streamline the workflow for QC calculation of explicitly solvated molecules. It automates the solvated-structure generation, force field fitting, configuration sampling, and the final extraction of microsolvated cluster structures that QC packages can readily use to predict molecular properties of interest. AutoSolvate is available through both a command line interface and a graphical user interface, making it accessible to the broader scientific community. To improve the quality of the initial structures generated by AutoSolvate, we investigated the dependence of solute-solvent closeness on solute/solvent identities and trained a machine learning model to predict the closeness and guide initial structure generation. Finally, we tested the capability of AutoSolvate for rapid data set curation by calculating the outer-sphere reorganization energy of a large data set of 166 redox couples, which demonstrated the promise of the AutoSolvate package for chemical discovery efforts.
@article{auto1,author={Hruska, Eugen and Gale, Ariel and Huang, Xiao and Liu, Fang},journal={J. Chem. Phys.},title={{AutoSolvate : A Toolkit for Automating Quantum Chemistry Design and Discovery of Solvated Molecules}},url={https://doi.org/10.1063/5.0084833},doi={10.1063/5.0084833},year={2022},preprint={https://chemrxiv.org/engage/chemrxiv/article-details/61db9b22db4d9f0ae2929eff},}
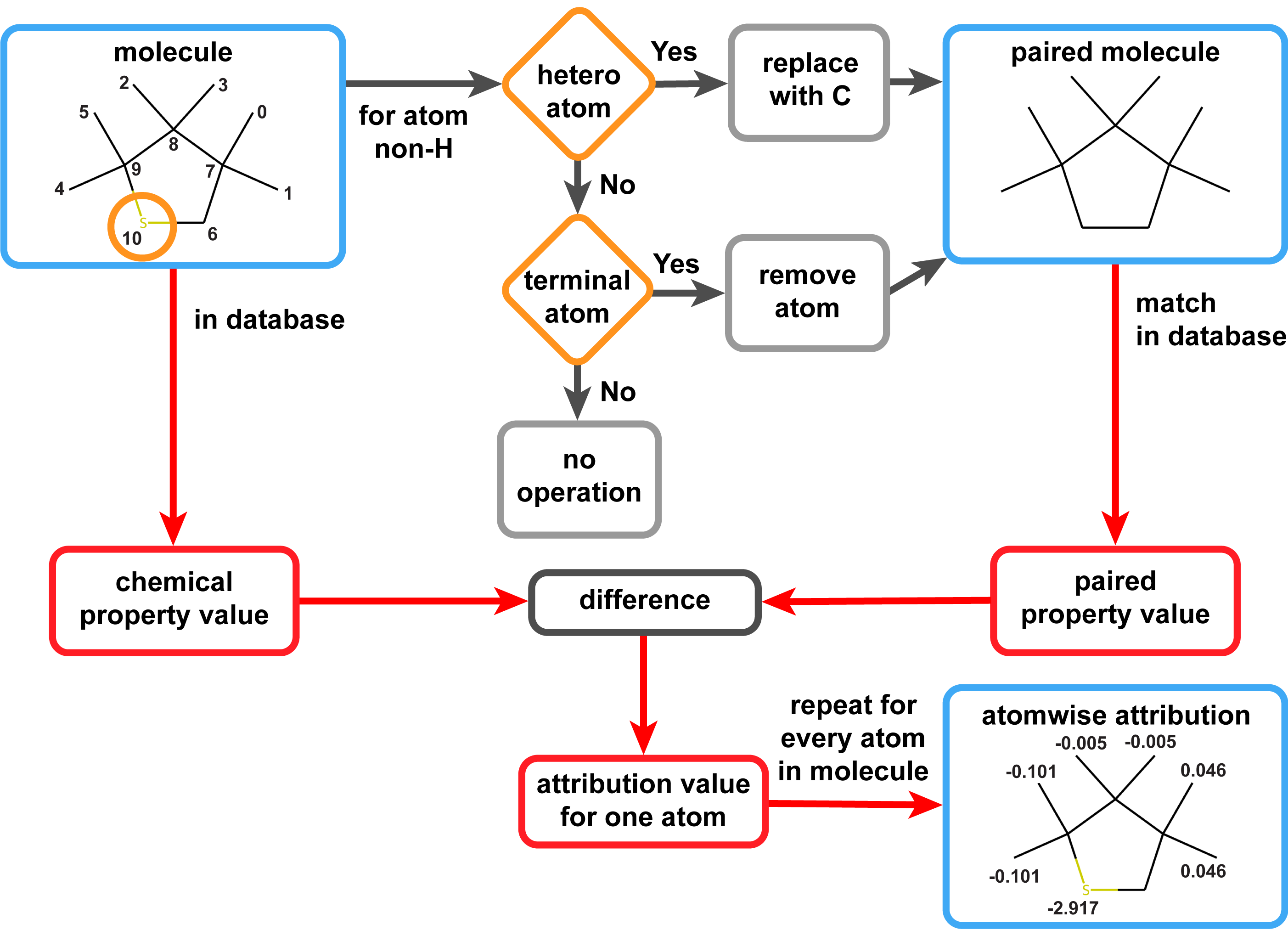
Ground truth explanation dataset for chemical property prediction on molecular graphs
Interpretation of chemistry on an atomic scale improves with explainable artificial intelligence (XAI). The parts of the molecule with the most significant influence on the chemical property of interest can be visualized with atomwise and bondwise attributions. Nonetheless, the attributions from different XAI methods regularly disagree substantially, causing uncertainty about which explanation is correct. To determine a ground truth for attributions, we define chemical operations which avoid alchemical steps or approximations and allow extracting one attribution per atom or bond from existing datasets of chemical properties. This general procedure allows for generating large datasets of ground truth attributions. The approach allowed us to create a ground truth explanation dataset with more than 5 million data points for the HOMO-LUMO gap chemical property. This open-source dataset of atomistic ground truth explanations may serve as a reference for XAI approaches.
2020
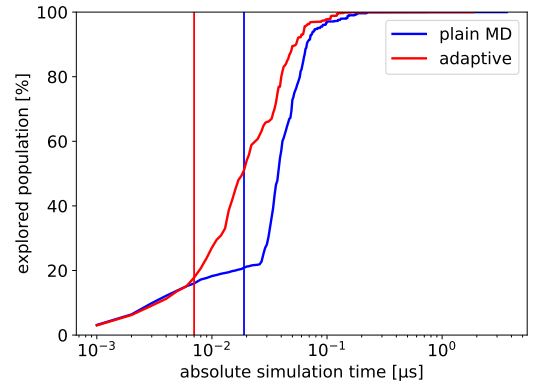
Extensible and Scalable Adaptive Sampling on Supercomputers
Eugen Hruska, Vivekanandan Balasubramanian, Hyungro Lee, and
2 more authors
The accurate sampling of protein dynamics is an ongoing challenge despite the utilization of high-performance computer (HPC) systems. Utilizing only “brute force” molecular dynamics (MD) simulations requires an unacceptably long time to solution. Adaptive sampling methods allow a more effective sampling of protein dynamics than standard MD simulations. Depending on the restarting strategy, the speed up can be more than 1 order of magnitude. One challenge limiting the utilization of adaptive sampling by domain experts is the relatively high complexity of efficiently running adaptive sampling on HPC systems. We discuss how the ExTASY framework can set up new adaptive sampling strategies and reliably execute resulting workflows at scale on HPC platforms. Here, the folding dynamics of four proteins are predicted with no a priori information.
@article{hruska2019extensible,title={{Extensible and Scalable Adaptive Sampling on Supercomputers}},author={Hruska, Eugen and Balasubramanian, Vivekanandan and Lee, Hyungro and Jha, Shantenu and Clementi, Cecilia},journal={J. Chem. Theory Comput.},year={2020},url={https://doi.org/10.1021/acs.jctc.0c00991},doi={10.1021/acs.jctc.0c00991},}
2018
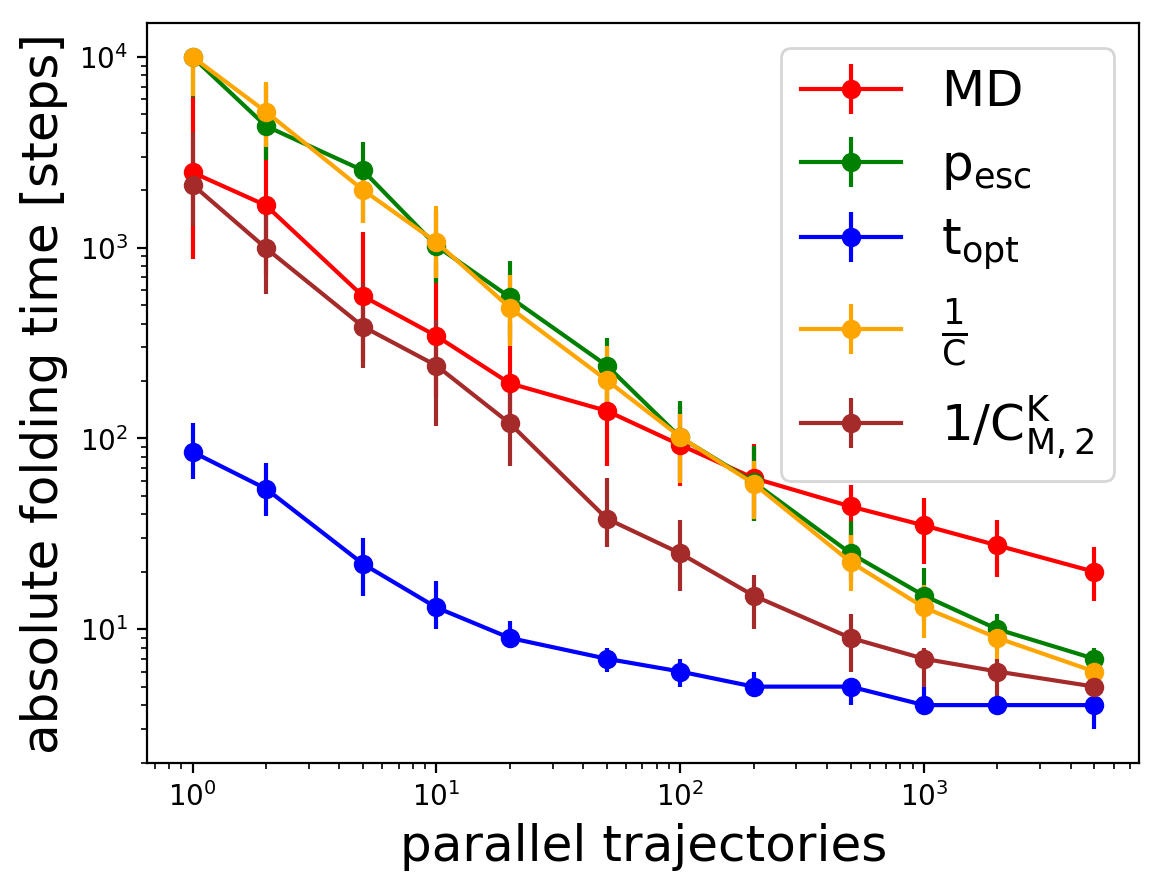
Quantitative comparison of adaptive sampling methods for protein dynamics
Eugen Hruska, Jayvee R Abella, Feliks Nüske, and
2 more authors
Adaptive sampling methods, often used in combination with Markov state models, are becoming increasingly popular for speeding up rare events in simulation such as molecular dynamics (MD) without biasing the system dynamics. Several adaptive sampling strategies have been proposed, but it is not clear which methods perform better for different physical systems. In this work, we present a systematic evaluation of selected adaptive sampling strategies on a wide selection of fast folding proteins. The adaptive sampling strategies were emulated using models constructed on already existing MD trajectories. We provide theoretical limits for the sampling speed-up and compare the performance of different strategies with and without using some a priori knowledge of the system. The results show that for different goals, different adaptive sampling strategies are optimal. In order to sample slow dynamical processes such as protein folding without a priori knowledge of the system, a strategy based on the identification of a set of metastable regions is consistently the most efficient, while a strategy based on the identification of microstates performs better if the goal is to explore newer regions of the conformational space. Interestingly, the maximum speed-up achievable for the adaptive sampling of slow processes increases for proteins with longer folding times, encouraging the application of these methods for the characterization of slower processes, beyond the fast-folding proteins considered here.
@article{hruska2018quantitative,title={Quantitative comparison of adaptive sampling methods for protein dynamics},author={Hruska, Eugen and Abella, Jayvee R and N{\"u}ske, Feliks and Kavraki, Lydia E and Clementi, Cecilia},journal={J. Chem. Phys.},year={2018},url={https://doi.org/10.1063/1.5053582},doi={10.1063/1.5053582},}